 |
|
(자료 :글로벌의약품 생산거점 인프라구축 방안연구, 식품의약품안전평가원) |
주주총회소집공고
| 2021 년 12 월 2 일 | ||
| 회 사 명 : | 프레스티지바이오로직스 주식회사 | |
| 대 표 이 사 : | 양 재 영 | |
| 본 점 소 재 지 : | 충청북도 청주시 흥덕구 오송읍 오송생명1로 197 (연제리) | |
| (전 화) 043-232-1552 | ||
| (홈페이지) http://www.prestigebiologics.com | ||
| 작 성 책 임 자 : | (직 책) 전 무 | (성 명) 임 영 수 |
| (전 화) 043-232-1552 | ||
주주총회 소집공고
| (2021년 1차 임시주주총회) |
주주님의 건승과 댁내의 평안을 기원합니다.
당사는 상법 제363조와 정관 제26조에 의하여 2021년 1차 임시주주총회를 아래와 같이 소집하오니 참석하여 주시기 바랍니다.
(상법 제542조의 4 및 정관 제26조의 2에 의거하여 발행주식총수의 1% 이하 소유주주에 대하여는 이 공고로 소집통지에 갈음하오니 참고하여 주시기 바랍니다)
- 아 래 -
1. 일시 : 2021년 12월 17일 금요일, 오전 10시
2. 장소 : 충청북도 청주시 흥덕구 오송읍 오송생명1로 197 (연제리), 2층 대강당
3. 회의 목적사항
부의사항
(1) 제1호 의안 : 정관 일부 변경의 건
(2) 제2호 의안 : 감사위원이 되는 사외이사 선임의 건
제2-1호 의안 : 사외이사 정진섭 선임의 건
제2-2호 의안 : 사외이사 나경아 선임의 건
(3) 제3호 의안 : 감사위원회 위원 선임의 건
제3-1호 의안 : 사외이사 김영준 선임의 건
제3-2호 의안 : 사외이사 정진섭 선임의 건
제3-3호 의안 : 사외이사 나경아 선임의 건
4. 경영참고사항 비치
「상법」제542조의4 제3항 의거, 주주총회 소집통지 또는 공고를 당사의 본점, 금융위원회, 한국거래소 및 한국예탁결제원 증권대행부에 비치 또는 공시하오니 참조하시기 바랍니다.
5. 전자투표 및 전자위임장 권유에 관한 사항
당사는「상법」제368조의4에 따른 전자투표제도와「자본시장과 금융투자업에 관한법률 시행령」제160조제5호에 따른 전자위임장 권유제도를 이번 주주총회에서 활용하기로 결의하였고, 이 두 제도의 관리업무를 삼성증권에 위탁하였습니다.
주주님들께서는 아래에서 정한 방법에 따라 주주총회에 참석하지 아니하고 전자투표방식으로 의결권을 행사하시거나, 전자위임장을 수여하실 수 있습니다.
가. 전자투표 및 전자위임장권유관리시스템
- 전자투표 및 전자위임장 위탁기관 : 삼성증권 (온라인 주총장)
- 인터넷, 모바일 주소 : https://vote.samsungpop.com
나. 전자투표 행사 및 전자위임장 행사기간
- 기간 : 2021년 12월 7일 오전 9시 ~ 2021년 12월 16일 오후 5시
- 투표기간 중 24시간 이용 가능
다. 인증서를 이용하여 시스템에서 주주본인 확인 후 의안별 전자투표 행사 또는
전자 위임장 수여
- 주주확인용 인증서의 종류: 코스콤 증권거래용 인증서, 금융결제원 개인용도
제한용 인증서 등
라. 수정동의안 처리
- 주주총회에서 상정된 의안에 관하여 수정동의가 제출되는 경우 기권으로 처리
- 한국예탁결제원 전자투표서비스 이용 약관 제11조 제2항
6. 주주총회 참석시 준비물
- 직접행사 : 참석장, 신분증
- 대리행사 : 참석장, 위임장 원본(인적사항 기재, 인감날인), 대리인 신분증,
주주의 인감증명서
I. 사외이사 등의 활동내역과 보수에 관한 사항
1. 사외이사 등의 활동내역
가. 이사회 출석률 및 이사회 의안에 대한 찬반여부
| 회차 | 개최일자 | 의안내용 | 사외이사 등의 성명 | |
|---|---|---|---|---|
| 김영준 (출석률: 100%) |
안영욱 (출석률: 71%) |
|||
| 찬 반 여 부 | ||||
| 21-1 | 2021.01.22 |
-오송 제2캠퍼스 신축공사를 위한 시공사 선정의 건 -전자투표 도입 승인의 건 -전자투표와 전자위임장에 대한 위탁계약 체결의 건 -관계사간 계약의 건 -코스닥상장을 위한 신주 모집 승인의 건 |
참석(찬성) | 참석(찬성) |
| 21-2 | 2021.02.04 | -코스닥상장을 위한 신주 발행 승인의 건 | 참석(찬성) | 참석(찬성) |
| 21-3 | 2021.02.25 | -정기주주총회 소집의 건 | 참석(찬성) | 참석(찬성) |
| 21-4 | 2021.04.19 | -신규시설 투자의 건 | 참석(찬성) | 참석(찬성) |
| 21-5 | 2021.06.10 | -신규시설 투자의 건 -자금 차입 검토의 건 |
참석(찬성) | 참석(찬성) |
| 21-6 | 2021.06.29 | -산업은행 시설자금 차입의 건 -채무보증 결정의 건 |
참석(찬성) | 불참 |
| 21-7 | 2021.11.05 | -국내 출장 규정 제정의 건 -관계사간 계약의 건 -임시주주총회 소집의 건 |
참석(찬성) | 불참 |
나. 이사회내 위원회에서의 사외이사 등의 활동내역
| 위원회명 | 구성원 | 활 동 내 역 | ||
|---|---|---|---|---|
| 개최일자 | 의안내용 | 가결여부 | ||
| - | - | - | - | - |
2. 사외이사 등의 보수현황
| (단위 : 백만원) |
| 구 분 | 인원수 | 주총승인금액 | 지급총액 | 1인당 평균 지급액 |
비 고 |
|---|---|---|---|---|---|
| 사외이사 | 2명 | 1,000 | 18 | 9 | - |
II. 최대주주등과의 거래내역에 관한 사항
1. 단일 거래규모가 일정규모이상인 거래
| (단위 : 억원) |
| 거래종류 | 거래상대방 (회사와의 관계) |
거래기간 | 거래금액 | 비율(%) |
|---|---|---|---|---|
| - | - | - | - | - |
2. 해당 사업연도중에 특정인과 해당 거래를 포함한 거래총액이 일정규모이상인 거래
| (단위 : 억원) |
| 거래상대방 (회사와의 관계) |
거래종류 | 거래기간 | 거래금액 | 비율(%) |
|---|---|---|---|---|
| - | - | - | - | - |
III. 경영참고사항
1. 사업의 개요
가. 업계의 현황
(1) 산업의 특성
(가) 산업의 동향
바이오의약품은 사람이나 다른 생물체에서 유래하는 세포, 단백질, 유전자 등을 원료로 하여 제조한 의약품으로서 보건위생상 특별한 주의가 필요한 의약품을 말하며, 성분에 따라 생물학적제제, 단백질의약품, 항체의약품, 세포치료제 및 유전자치료제 등으로 구분하고 있습니다.
바이오의약품은 합성의약품과 비교하여 약의 체내 분해시 발생하는 대사 산물이 없어 독성이 낮고, 질환의 발병기전과 관련된 체내 단백질에 선택적으로 작용한다는 장점을 가지고 있습니다. 다만 살아있는 세포에서 만들어지는 큰 단백질로 이루어져 있기 때문에 일반적으로 크기가 훨씬 작은 저분자 화합물(small molecule)로 구성되는 합성의약품보다 200~1,000배 가량 크고 구조적으로 복잡하여 제조 및 보관 조건에 민감하다는 특성을 가지고 있습니다. 치료상 장점과 생산 및 보관의 까다로움으로 인하여 합성의약품에 비하여 바이오의약품 분야가 상대적으로 많은 시간과 자금이 소요되며, 높은 진입 장벽을 가지고 있어 더 높은 시장가격을 인정받고 있습니다.
|
구 분 |
합성의약품 |
바이오의약품 |
|
원료 |
합성화학물질 |
생물체 유래물질 (세포, 조직, 유전물질 등 ) |
|
원료의 고려사항 |
품질(시험분석으로 확인가능) |
시험분석으로 확인 가능한 품질 외에 공여(기증)자의 동의 등의 윤리성, 감염 질환 확인 등의 안정성 확보 필요 |
|
구조 |
물리화학적 특성이 명확한 저분자 구조 |
정확한 특성 분석이 불가능하고, 활성과 구조가 일정하지 않음 |
|
제품의 안정성 |
대부분 온도·빛 등 환경에 안정적 |
온도·빛·pH 등 외부환경에 민감하며 미생물 오염에 취약 |
|
대부분 36개월 |
(세포 치료제 사례) 대부분 3일이내 (유전자 치료제 사례) 영하 135도에서 24개월 |
|
|
제조 |
간단한 화학적 합성으로 대량생산 |
복잡한 제조과정의 맞춤형 소량생산 |
|
원료, 공정, 설비변화가 품질에 미치는 영향이 비교적 적음(제조공정의 변이성이 매우 낮음) |
원료, 공정, 설비의 변화가 의약품 자체를 변화시킴(제조공정에 따른 변이성 매우 높음) |
|
|
상대적으로 복제가 쉽고 낮은 제조비용 |
유사 복제만 가능하고 높은 제조비용 |
|
|
치료효과 |
비교적 명확한 약리기전, 불특정 다수에 일관적 효과 기대 |
세포 치료제: 약리기전이 불확실 유전자 치료제: 복합적인 기전 환자에 따른 맞춤형 치료 가능 |
|
대부분 질병의 증상개선에 그침 |
질병의 근본적인 원인치료 가능 |
|
|
투약방법 |
대부분 경구·주사 등 일반적 투여경로 |
대부분 주사 또는 주입투여하나, 이식 등 시술을 통하여 투여하기도 함 |
(자료:바이오의약품 산업동향보고서(2020.12),한국바이오의약품협회,회사 일부수정)
합성의약품은 다른 제조과정을 통해서도 성분과 구조가 원약과 동일한 복제약의 제조가 가능한 반면, 바이오의약품은 일반적으로 합성의약품에 비해 크기가 크고 복잡한 고분자 구조를 가지고 있으며, 생물체를 이용하여 복잡한 제조공정을 거쳐야 되므로 배양기술과 환경, 방법에 따라 단백질의 변형, 단백질의 당화수준이 달라져 원약과 비교하여 성분과 구조가 동일하지 않은 물질이 만들어지기 때문에 생물학적 동등성을 갖으면서 구조적으로 유사한 복제가 쉽지 않습니다.
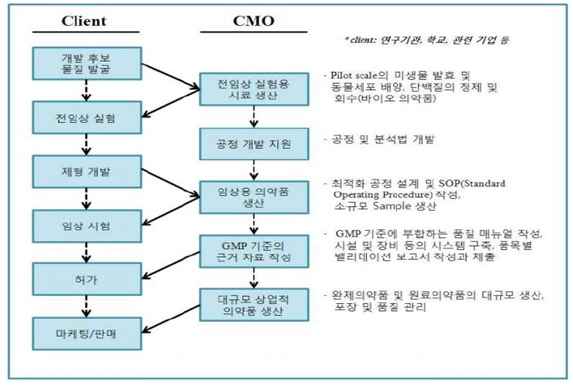
바이오의약품 산업의 가치사슬(Value Chain)은 연구개발, 생산, 운송과 유통 그리고 판매라는 다양한 단계로 구분되고 각 단계는 매우 복잡하고 동시에 대규모 투자를 필요로 하므로 몇몇 소수의 다국적 거대 제약 기업을 제외하고 전 기능을 단독으로 수행하기 어렵습니다. 바이오의약품의 생산을 위해서는 대규모 투자가 필요할 뿐만 아니라, 플랜트 및 엔지니어링 설계, 건설, 밸리데이션, 안정화 등에 최소 3년 이상의 시간이 필요합니다. 또한, 동물세포를 이용하는 세포 배양, 정제, 충전 등 생산 전 과정에서 GMP(Good Manufacturing Practice)에 부합하는 높은 수준의 품질관리 역량이 필수적으로 요구됩니다.
이에 따라, 바이오의약품은 합성의약품과 달리 생산공정이 복잡하여 전문적인 CMO의 필요성이 더욱 커지고 있습니다. CMO는 산업 초기에 제약사들이 내부 제조 시설(in-house manufacturing)만으로 시장 수요를 충족시킬 수 없을 때, 추가적인 제조 역량을 제공하던 것에서 출발하여, 생산공정 기준 및 시험법 개발확인, 제품제형 개발지원, 해외등록서류 개발 등을 포함하여 업무범위가 확장되었습니다.
|
|
|
(자료 :글로벌의약품 생산거점 인프라구축 방안연구, 식품의약품안전평가원) |
한편, 제약사들은 개발중인 의약품의 등록을 위하여 전용 제조 시설을 설치하였지만 임상 3상 단계에서 실패하는 사례를 종종 볼 수 있습니다. 이 경우, 특정 의약품을 위하여 설치한 제조 시설을 더 이상 필요하지 않게 되는 투자 위험이 있으며, 투자위험을 줄이기 위해 아웃소싱 제조에 대한 수요가 지속적으로 증가하고 있습니다. 이에 연구개발의 위험성 극복 및 제한된 자원의 효율적인 활용을 위하여 대기업 뿐만 아니라 중소기업도 개방형 혁신(Open Innovation)전략을 추구하게 되면서, 신약개발 사업모델이 신약개발에서 생산 및 영업까지 모든 단계를 수행하는 완전통합형(FIPCO, Fully Integrated Pharma Co.)에서 신약개발의 가치사슬(Value Chain)별로 전문·세분화되어 수행하면서 가상적으로 통합하여 수행하는 가상통합형(VIPCO, Virtually Integrated Pharma Co.) 모델로 진화하고 있습니다. (세계의약품 산업 및 국내산업경쟁력 현황, 한국수출입은행 해외경제연구소, 2017.08)
이러한 추세로 인하여 과거에는 대형 제약사가 대형 CMO와 계약을 맺는 시장이 주를 이루었으나 최근에는 중견 제약사, 바이오제약사, virtual 제약사 등과 계약을 맺는 중ㆍ소규모 또는 신흥 CMO가 새로운 기회를 얻는 등 산업 생태계의 변화가 나타나고 있으며, 산업 초기 전임상 물질, 임상 물질, 상업화에 위탁 생산만 담당하던 CMO 시장에서도 단순 생산 이상의 업무범위를 갖는 차별화된 CMO가 나타나고 있습니다.
특히, 바이오시밀러 생산의 경우 대규모 생산시설 및 수율이 중요한 이슈이며, 일반적인 제품 생산보다 훨씬 다양한 노하우와 기술이 존재하고, 이를 활용한 생산 공정 기술 확립이 바이오시밀러 생산에서 매우 중요한 역할을 차지합니다. 바이오시밀러에 대한 관심과 수요가 증가함에 따라 대규모 생산시설과 고난도의 생산기술을 필요로 하는 CMO 시장도 함께 성장하고 있으며, CMO 산업 내에서도 경쟁력을 확보하기 위해 다양한 방면으로 연구 및 개발이 진행되고 있습니다. 이에, 배양 배지 선정, 공정개발, 세포주 개발, Scale up, 기술 이전, 시험 분석법 개발 등의 실제 개발 업무에도 참여하는 CDO(Contract Development Organization)로서의 역할이 결합된 CDMO(Contract Development Manufacturing Organization)사업이 부상하고 있습니다.
(나) 산업의 특성
바이오의약품 위탁생산사업(Contract Manufacturing Organization)은 실험용 시료생산에서부터 공정개발지원, 임상 및 완제의약품 생산에 이르기까지의 전문적인 서비스를 제공하는 사업으로 제약산업의 세계화와 산업 자체의 급속한 성장, 기업 규모 증대 및 여러 사회·정책적 변화 등으로 활성화되고 있습니다.
바이오의약품은 생물체를 이용하여 복잡한 제조공정을 거쳐야 하고, 인간의 생명과 보건에 관련된 제품을 생산하는 국민의 건강과 직결된 산업으로서 제품의 개발에서 임상시험, 인허가 및 제조, 유통 판매 등 전 과정이 국가에 의해 엄격히 규제되는 산업입니다. 화학(Chemical) 제품은 대규모 생산이 용이하고 품질의 균일성, 약효발현 등이 장점으로 의약품산업의 성장을 이끌어왔으나, 의약품산업이 고도화됨에 따라 화학적 합성을 통한 신물질 개발의 한계가 도래하고 있습니다. 반면, 바이오의약품의 경우 생산 노하우(Know-how) 및 시설의 Validation 등까지 수년간의 시간과 막대한 시설투자가 선행되어야 하고 호르몬, 항체, 백신 및 생체기능성 단백질을 살아있는 미생물/동물숙주세포에서 대량생산, 추출하여 의약품으로 개발하는 첨단 생명공학 기술이 요구되며, 각 국의 GMP시설 운영 및 품질관리 능력이 필요한 고부가가치 산업입니다.
GMP 규정은 의약품 개발 및 상업화에 이르기까지 준수해야하는 규정으로 임상시험에 사용하는 의약품부터 GMP기준에 맞게 제조하고 각국의 임상시험계획승인 신청시 GMP 기준에 맞게 제조되었음을 증명하는 자료를 제출해야 합니다. 각 현지의 GMP 기준에 대한 경험 및 능력을 갖추기 위해 많은 시간이 필요하므로 이미 가격경쟁위주의 범용화가 진행된 합성의약품 분야보다는 상대적으로 바이오의약품 CMO산업은 기술 및 진입장벽은 높은 편이라 할 수 있습니다.
당사는 제1공장 준공 후 7개월만인 2019년 1월, 주요 생산장비 및 생산지원설비의 설치·운전 적격성 평가부터 우수의약품 제조관리기준(GMP, Good Manufacturing Practice) 인증 획득을 위한 공정 밸리데이션 (PV) 배치 생산을 한번의 실패없이 성공하였고, GMP실사를 완료하여 2019년 4월 식품의약품안전처로부터 의약품제조 및 품질관리기준 (KGMP) 적합판정을 받았습니다.
(2) 산업의 성장성
(가) 시장의 성장요인
① 세계 의약품 시장에서 바이오의약품 비중의 증가
합성의약품은 합성신약의 성공빈도가 낮아져 R&D투자효율성이 낮아지고 있는 반면, 바이오의약품은 합성의약품 대비 독성이 낮아 부작용이 적고, 표적 단백질 혹은 장기에 직접적인 효능을 발휘하여 우수한 효과가 있으며, 생명공학기술 발전 등으로 성공확률이 높아 바이오의약품 시장이 급성장하고 있습니다
전체의약품에서 바이오의약품이 차지하는 비중은 2012년 20%에서 2019년 29%로 증가하였으며, 2026년 35%로 증가할 것으로 전망됩니다. 또한, 2019년 글로벌 상위 100대 의약품 중 바이오의약품 비중이 53%로 절반 이상을 차지하는 것으로 추정됩니다(Evaluate pharma(2020)). 바이오의약품 시장의 성장과 함께 바이오의약품 위탁제조 및 연구개발시장의 성장도 함께 이루어질 것으로 예상합니다.
② 바이오의약품 개발사들의 비용효율 및 투자리스크 관리
적절한 생산설비를 보유하지 못한 제약기업이 새로운 바이오의약품 출시를 준비중이라면 허가 받기 2~4년 전부터 큰 위험 부담을 감수해야 합니다. 신약이 100% 허가를 받을 수 있는 것이 불확실한 상황에서 공장 건설에 수년이 걸리고 대규모 투자자금이 소요됩니다. 따라서, 위탁생산업체(CMO)에 위탁생산을 함으로써, 공장 건설에 필요한 높은 초기 투자 비용이 소요되지 않고, 시간을 절약하고 위험부담을 낮출 수 있습니다.
한편, 2018년 12월 말 기준 미국 FDA 허가 신약은 59개로 역대 최고치를 경신했으며, 그 중 약 30%인 17개가 바이오 신약으로 나타났습니다. 생명공학의 발전으로 바이오신약 개발증가와 리스크 관리 측면에서의 CMO의 수요도 함께 증가될 것으로 예상됩니다.
③ 공급안정화 및 경쟁력향상을 위한 대형제약기업의 아웃소싱 증가
의약품의 경우 생산지를 변경하려면 추가적인 허가절차가 필요하여 긴 시간이 소요되는데, CMO를 포함하여 복수의 공장에 대해 사전에 허가작업을 진행해 놓으면 수요 급증이나 천재지변 등으로 인한 제품 부족에 대응할 수 있습니다. 또한, 생산지 등을 변경할 경우 허가절차 등에 추가 시간이 소요되어 의약품 허가전쟁에 불리한 점으로 작용될 수 있기 때문에 생산역량을 갖춘 대형제약기업의 경우에도 아웃소싱을 적극 활용하고 있습니다. 또한 최근 미국ㆍ유럽에서 COVID-19 확산으로 인한 의약품 생산중단 리스크를 줄이기 위해 듀얼소싱(Dual Sourcing)수요가 증가하여, 자체적인 생산에 아웃소싱 CMO를 추가하거나, 두 군데 이상의 CMO 업체에 위탁생산을 맡기는 경우도 발생하고 있습니다.
④ 의약품 개발 전단계에 대한 아웃소싱의 수요 증가
바이오 제약기업들이 효율적인 연구개발(R&D)을 위해 학계 및 임상대행기업과의 제휴가 증가하고 있으며, 연구 및 생산시설이 없거나 부족한 중소규모의 기업들 뿐만 아니라 대기업들도 리스크를 낮추기 위해 아웃소싱 서비스에 대한 수요가 증가하고 있습니다. 이에 글로벌 CRO시장 또한 2016년~ 2021년 연평균 12.8%로 성장할 것으로 전망되고 있으며(세계 바이오의약품 산업 동향 및 전망, 2019), 해당 산업과 함께 이루어지는 의약품 상업화를 위한 세포주 및 공정개발 등을 포함하는 개발업무에 대한 아웃소싱 시장도 성장할 것으로 예상하고 있습니다.
⑤ 특허만료로 인한 바이오시밀러 시장 확대
바이오시밀러 시장은 바이오신약에 비해 상대적으로 개발비용 및 개발기간을 절감할 수 있고, 제품가격이 바이오신약보다 저렴하여 세계 각국의 의료비 재정부담을 축소하고 의약품에 대한 환자의 접근성 개선하여 도입이 적극적으로 장려되고 있습니다. 최근 레미케이드(Remicade), 리툭산(Rituxan), 허셉틴(Herceptin), 란투스(Lantus) 등의 항체 블록버스터 의약품의 특허가 만료되며, 바이오시밀러 시장이 개화되었습니다. 향후 10년 내에 키트루다(Keytruda), 옵디보(Opdivo), 스텔라라(Stelara) 등의 블록버스터 신약 물질들의 특허 만료가 예정되어 있는 점은 CMO 업체들에 긍정적인 요소로 작용할 것입니다. 최근 특허가 만료된 주요 블록버스터 항체 신약들의 2018년 합산 매출액은 $39bn(약 46조원)이며, 향후 특허 만료 예정인 주요 블록버스터 항체 약물들의 2018년 합산 매출액은 $34bn(약 40조원)입니다. 2024년 키트루다, 옵디보, 스텔라라 등의 고성장으로 합산 매출이 $58bn(약 69조원)인 점을 감안하면 향후 특허 만료에 따른 시밀러 시장 확대로 CMO의 성장은 계속될 수 있을 것으로 전망되고 있습니다(Cortellis, 키움증권보고서 일부인용).
⑥ 규제 등 정책적 측면
FDA 등에서 제시하는 의약품 제조 및 품질관리기준의 강화로 인해 바이오의약품을 연구 ·개발하는 제약사들이 관련 기준을 충족하는 생산시설을 자체적으로 갖추기는 어렵게 된 반면, 지적재산권 보호 강화 정책에 따라 충분한 기술이전을 통하여 위탁생산의약품의 품질에 대한 신뢰성이 담보될 수 있게 되었습니다. 또한, FDA 등의 의약품 규제기관에서는 의약품의 안정적인 공급을 확보할 수 있도록 복수의 생산라인을 운영하도록 권고하고 있습니다. 이로 인하여 자체적으로 복수의 생산라인이 없는 회사들이 CMO를 이용하는 경우가 많아지고 있습니다.
(나) 시장규모 및 전망
글로벌 바이오의약품 시장규모와 전망은 조사기관과 발표시점에 따라 차이가 있으나, Evaluate pharma(2020)’에 따르면 2019년 기준 전체 의약품 시장은 9,100억 달러이며, 바이오의약품 시장은 2,660억 달러로 전체의약품 대비 29%를 차지하고 있습니다. 최근 8년(2012~2019년)간 연평균 8.6%로 성장하였으며, 향후 7년 (2020~2026년)간 연평균 10.1% 성장하여 2026년 5,050억 달러에 달할 것으로 전망하고 있습니다.
바이오의약품 산업동향보고서(한국바이오의약품협회, 2020)에 따르면, 2019년 글로벌 매출 상위 10위 의약품중 바이오의약품은 8품목이고, 상위 1위는 휴미라(Humira, Abbvie사)로 192억 달러를 기록하였으며, 아바스틴(Avastin, Roche사)은 6위로 71억달러, 허셉틴(Herceptin, Roche사)은 9위로 약 61억달러를 기록하였습니다(Nature Reviews, 2020).
한편, 당사의 주제품으로 생산을 예정하고 있는 바이오시밀러 시장의 경우, 2017~2023년 연평균 30.6%로 고성장하여 2017년 97억달러에서 2023년 481억달러에 이를 것으로 전망됩니다(Frost & Sullivan 2019, 생명공학정책연구센터). 또한, 2019년 기준 글로벌의약품 R&D투자는 1,860억달러로 추정하고 향후 7년간(2019~2026년) 연평균 3.2% 증가하여 2026년에는 2,325억달러에 이를 것으로 전망하고 있습니다.
바이오의약품의 시장규모 및 R&D투자의 증가는 개발사들의 비용효율 및 투자리스크 관리, 공급안정화 및 성장경쟁력 집중전략 등으로 인해 연구개발 및 제조의 아웃소싱 수요의 증가로 이어지고 있으며, Frost & Sullivan에 따르면 글로벌 바이오 CDMO 시장규모는 2019년기준 119억 달러이며, 향후 10년간 약 13.4%로 성장하여, 2025년 기준 약 253억달러의 시장이 형성될 것으로 전망하고 있습니다.
(3) 경기변동의 특성
(가) 경기변동 및 계절성
바이오의약품 산업은 생명 및 건강과 직접적인 연관이 있는 의약품을 연구ㆍ제조하는 산업으로 경기변동과 계절적요인에 따른 민감도가 비교적 낮아 타 산업에 비하여 비탄력적인 특성을 가집니다.
(나) 대체시장의 현황
의약품은 각 국가별 기준에 의거한 허가절차를 거쳐 승인을 받아야만 시판할 수 있고, 전문 의약품의 경우 의사의 처방전에 의해서만 약을 구입할 수 있도록 제도적으로 보호되어 있으므로 타 산업과 비교하여 대체시장의 형성이 쉽지 않은 특성을 지니고 있습니다.
다만, 바이오의약품 위탁서비스산업의 측면에서는 자체적으로 생산설비 및 개발역량을 확보하여 제조 및 연구개발을 진행하는 개발사가 될 수도 있지만, 대규모 생산시설 구축 및 전문 인력확보에 어려움이 있거나, 비용 효율 및 연구개발 집중 등을 원인으로 중ㆍ소형 제약사 뿐만 아니라 대형제약사에서도 아웃소싱 수요가 증가하는 추세입니다.
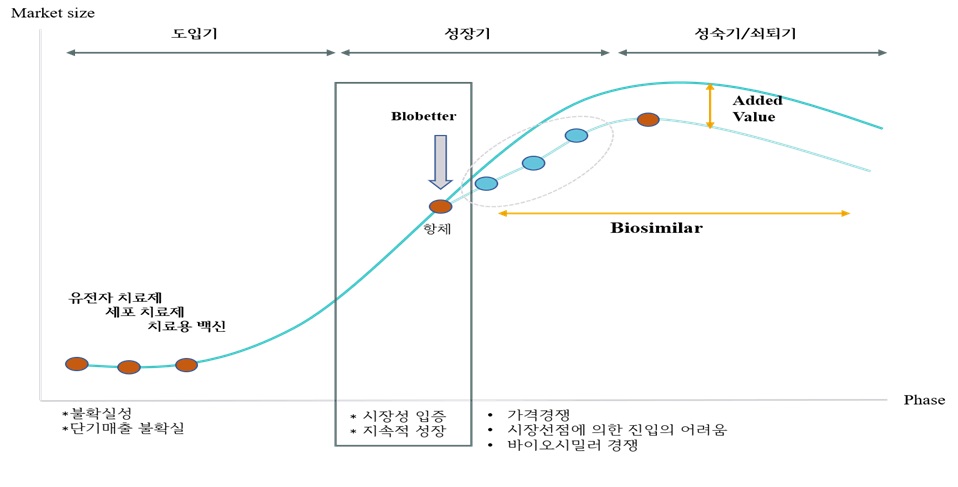
(다) 라이프사이클
[바이오의약품의 성장주기]
 |
|
(자료: 생명공학연구센터, 한국투자증권리서치센터) |
바이오의약품 시장의 라이프사이클은 오리지널 의약품의 특허보호로 인해 타 산업에 비하여 주기가 길고 안정적입니다. 오리지널 의약품의 특허 만료 이후, 바이오시밀러가 개발되어 시장에 진입하게 되면서, 의약품 가격의 하락과 수요의 증가로 처방받는 환자수가 증가하게 되며, 이로 인하여 지속적으로 성장하는 흐름을 보여주고 있습니다. HD201(허셉틴 바이오시밀러) 및 HD204(아바스틴 바이오시밀러)는 전세계 시장에 대해서 특허가 만료되기 시작하여 전체 바이오의약품의 성장 주기에서 초기 성숙기 단계에 해당됩니다. 바이오의약품 위탁서비스사업의 라이프사이클 또한 제품별로 5년~10년의 라이선스 및 공급계약을 체결하고, 기술이전 및 제조소 변경에 긴 시간과 비용이 소요되기 때문에 주기가 긴 편이며, 안정적인 라이프사이클을 유지하고 있습니다.
(4) 경쟁상황
2020년 7월 한국바이오의약품협회가 발간한 보고서에 따르면 국가별 바이오의약품 생산규모는 다음과 같습니다.
|
순위 |
국가 |
생산능력(리터) |
순위 |
국가 |
생산능력(리터) |
|
1 |
미국 |
1,791,326 |
5 |
아일랜드 |
190,000 |
|
2 |
한국 |
338,850 |
6 |
스위스 |
137,225 |
|
3 |
독일 |
268,850 |
7 |
프랑스 |
128,100 |
|
4 |
싱가포르 |
193,200 |
세계 |
3,708,561 |
|
(자료: 보건복지부,2018)
현재 자체설비를 가지고 있는 개발사 및 위탁제조사들이 성장하는 바이오의약품 수요에 대응하기 위해 인수합병 및 공장증설 등을 추진하여 생산능력확보에 많은 노력을 기울이고 있어 세계적인 생산규모는 더욱 커질 것으로 예상됩니다.
Bioplan Internal data(2019)에 따르면 전세계적으로 운영되고 있는 바이오의약품 제조시설은 1,813개로 추정됩니다. 연구개발과 초기 임상시험 시료를 제조하는 500L이하의 소규모 생산시설은 751개로 40% 이상을 차지하고 있으며 상업용 제품을 생산하는 10,000L이상의 대규모 생산 시설은 292개로 약 16%를 차지하고 있습니다. 또한, 전체 바이오의약품 제조시설 중에서 497개의 제조시설이 CMO 시설로 운영되고 있으며, 전체 제조시설 중에서 약 27%를 차지하고 있습니다. 일반적으로 전체 제조 시설 중에서 약 3분의 1이 상업용 제품을 생산하고 있는 것으로 추정됩니다.
한편, 규모에 따라서 CMO를 small, mid-sized, large로 구분할 수 있습니다.
- Small CMOs : 물질의 개발, 전임상 단계, 이른 단계의 임상용 시료를 생산하기 위하여 위탁하고 있습니다.
- Mid-sized CMOs : 2,000-12,000L 규모의 생산 Capacity를 보유하고 있는 CMOs를 통칭하며, 연간 30~150백만 달러의 매출이 발생하고 있습니다.
- Large CMOs : Stainless steel 방식 기반의 10,000L 이상의 배양기를 보유하고 있으며, 일반적으로 연간 150백만 달러 이상의 매출이 발생하고 있습니다.
제2공장까지 확보될 경우 Single use 와 Hybrid 방식(Single-use 방식과 stainless steel 방식이 조합된 Alita)을 통한 104,000L 규모 수준의 Large-sized CMO 업체로 분류될 것으로 예상하고 있습니다.
한편, 당사가 우선제조권한을 보유하고 있는 프레스티지바이오파마의 허셉틴 바이오시밀러 및 아바스틴 바이오시밀러의 경쟁상황은 다음과 같습니다.
① 허셉틴 바이오시밀러 개발현황
허셉틴 바이오시밀러의 경우 프레스티지바이오파마를 포함한 9개의 경쟁사가 임상 개발 이상의 단계에 있으며, 이 중 7개 의약품이 규제 당국의 판매 승인이 완료 또는 진행 중에 있습니다. 당사는 2022년 상업용제품제조에 따른 매출을 예상하고 있습니다.
|
번호 |
개발사 |
상품명 |
유통파트너사 |
개발단계 |
출시일 |
|
1 |
마일란(미국), 바이오콘(인도) |
Ogiviri® |
마일란(미국) |
EMA승인(2018) FDA승인(2017) |
유렵 2019.08 미국 2019.12 |
|
2 |
삼성 바이오에피스 (한국) |
Ontruzant® |
대웅제약, 머크(미국) |
EMA승인(2017) FDA승인(2019) |
유럽 2018.03 미국 2020.04 |
|
3 |
셀트리온(한국),테바(미국) |
Herzuma® |
테바(이스라엘), 먼디파마(EU) |
EMA승인(2018) FDA승인(2018) |
유럽 2018.05 미국 2020.03 |
|
4 |
엘러간(미국),암젠(미국) |
Kanjinti® |
암젠(미국) |
EMA승인(2018) FDA승인(2019) |
유럽 2018.06 미국 2019.07 |
|
5 |
화이자(미국) |
Trazimera® |
호스피라(미국) |
EMA승인(2018) FDA승인(2019) |
유럽 2019.04 미국 2020.02 |
|
6 |
상하이 헨리우스(중국) 어코드(영국) |
Zercepac® |
- |
EMA승인(2020) |
미정 |
|
7 |
Prestige Biopharma (싱가포르) |
Tuznue® |
먼디파마&알보젠 등 글로벌 제약회사 |
EMA신청(2019) FDA신청 예정 |
미정 |
|
8 |
Tanvex Biopharma Inc |
TX-05 |
- |
임상3상 |
미정 |
|
9 |
Gedeon Richter Ltd (헝가리) |
미정 |
Qilu제약(중국) |
임상3상 |
미정 |
주1) 개발단계 내용 중 괄호 안은 감독기관 승인 또는 신청연도
(자료: GlobalData, 각사 공시자료 및 발표자료)
② 아바스틴 바이오시밀러 개발 현황
아바스틴은 2004년 판매허가 이후 최근까지 글로벌 매출 10위권에 드는 블록버스터 의약품으로 전이성 대장암, 비소세포폐암 등의 적응증을 보유하고 있습니다. 아바스틴의 미국 특허는 2019년 7월 만료되었으며, 유럽의 경우 2022년 1월 만료가 예정되어 있습니다. 프레스티지바이오파마의 HD204(아바스틴 바이오시밀러)는 아바스틴의 유럽 특허가 만료되는 시점인 2022년 출시를 목표로 하고 있으며, 당사의 제조매출 또한 2022년 발생할 것으로 예상하고 있습니다.
|
번호 |
개발사 |
상품명 |
유통파트너 |
개발단계 |
출시일 |
|
1 |
엘러간(미국) |
Mvasi® |
암젠(미국) |
EMA승인(2018) FDA승인(2017) |
미국 2019.07 유럽 2022 예상 |
|
2 |
화이자(미국) |
Zirabev® |
- |
EMA승인(2019) FDA승인(2019) |
미국 2020.01 유럽 2022 예상 |
|
3 |
삼성바이오에피스(한국) |
Aybintio® |
머크(미국) |
EMA승인(2020) FDA신청(2019) |
미정 |
|
4 |
Centus Biotherapeutics |
Equidacent® |
- |
EMA승인(2019) |
미정 |
|
5 |
바이오콘(인도) |
Krabeva® |
마일란(미국) |
FDA 신청(2020_ |
미정 |
|
6 |
Prestige Biopharma (싱가포르) |
VasfordaTM |
- |
임상3상 |
미정 |
|
7 |
Innovent Biologics (중국) |
IBI305 |
- |
임상3상 |
미정 |
|
8 |
셀트리온(한국) |
CT-P16 |
- |
임상3상 |
미정 |
|
9 |
Outlook Therapeutics (미국) |
LYTENAVA™ |
비로프로(미국) |
임상3상 |
미정 |
|
10 |
Bio-Thera Solutions Ltd(중국) |
BAT1706 |
- |
임상3상 |
미정 |
|
11 |
닥터 레디 연구소(인도) |
VersavoTM |
- |
임상1상 |
미정 |
|
12 |
아포바이오로직스(캐나다) |
ABX-BEV |
- |
임상1상 |
미정 |
|
13 |
Tanvex BioPharma Inc (대만) |
TX16 |
- |
임상1상 |
미정 |
|
14 |
Zhejiang Teruisi Pharmaceutical (중국) |
TRS003 |
- |
임상1상 |
미정 |
주1) 개발단계 내용 중 괄호 안은 감독기관 승인 또는 신청연도
(자료: GlobalData, 각사 공시자료 및 발표자료)
(5) 자원조달의 특성
바이오의약품 CMO 사업의 경우 고객사 제품의 전체공정에 대한 기술을 이전받아 생산되어야 하고 각 제품별로 해당지역을 관할하는 규제당국의 허가를 받아야 합니다. 이에 제품별로 지정된 원부재료를 사용하여야 하며, 해당 원부재료의 구매비용 및 일정운영비를 더하여 환급받고, 경우에 따라 특정 설비 및 기계장치가 필요한 경우, 해당 장치의 설치 및 구매비용도 고객사로부터 환급 또는 지원받아 위탁생산하는 경우도 있습니다. 일반적으로는 각 현지의 CMO가 직접 고객사가 지정하는 원재료의 판매를 담당하는 현지업체와 접촉하여 계약하며, CMO는 현지업체와 대량 및 장기계약을 통해 안정적으로 원재료를 조달 받는 구조입니다.
(6) 관련 법령 또는 정부규제
바이오의약품은 각국의 규제기간으로부터 품목별로 제조허가를 별도로 받아야 하며,품목허가 시(시판허가 시) 제조소 현장실사를 통해 각 규제당국의 GMP 규정에 맞는 검증을 거쳐 제조허가를 승인 받을 수 있습니다.
GMP(Good Manufacturing Practice)는 우수한 의약품을 제조하기 위하여 공장에서 원료의 구입부터 제조, 출하 등에 이르는 모든 과정에 필요한 관리기준을 규정한 것입니다. 미국 FDA가 1963년 GMP를 제정ㆍ공표하면서 WHO(세계보건기구)와 각국에서 GMP를 도입하기 시작했고, 한국에도 1977년에 처음 도입되어 1995년 의무화되었습니다.
|
국가/국제기구 GMP |
|
|
cGMP |
미국 식품의약청(FDA)가 만든 가장 높은 수준의 GMP |
|
EU GMP |
유럽의약품청(EMA)에서 정한 가이드라인 |
|
KGMP |
한국 식품의약품안전처(MFDS)의 우수의약품제조 및 품질관리기준 |
|
WHO GMP |
WHO 주관 입찰을 통해 수출하는 백신, 생물학 제제 제조관리 기준 |
|
PIC/S GMP |
의약품 상호실사협력기구의 GMP 가이드라인이며 회원국간 의약품 실사정보를 공유해 의약품 허가 등록시 필요한 GMP 실사를 면제 또는 간소화할 수 있음 |
(자료: 한미약품 홈페이지, 회사재구성)
당사는 2019년 4월 식품의약품안전처로부터 의약품 제조 및 품질관리기준 적합판정을 획득(KGMP) 하였으며, 3년 이내 바이오의약품 전문수탁 제조업체 제조 및 품질관리기준 실시상황 평가를 진행하여 GMP에 대한 관리감독을 받을 예정입니다. 또한 유럽 및 미국을 포함한 글로벌 판매를 위하여 각국의 의약품 품질관리 기관(EMA, FDA 등)으로부터 GMP(EU GMP, cGMP 등)를 획득하여야 합니다. 당사는 2018년 11월 유럽에서의 임상 의약품 사용 승인을 위한 GMP 실사 제도인 EU QP (Qualified Person) 실사를 성공적으로 마쳤습니다.
2019년 4월 당사의 관계사인 프레스티지바이오파마는 HD201(허셉틴바이오시밀러)의 EMA 품목허가(판매허가)를 신청하였습니다. 품목허가 절차는 9단계로 이루어지며 기간은 통상 1년 정도 소요됩니다. 허가절차의 하나로 2020년 1월 27일부터 EU GMP실사를 진행하였으며, 현재 코로나바이러스감염증-19(COVID-19)으로 인하여 EMA 측의 실사연기요청으로 연기되었으며 이후 일정에 대한 자세한 사항은 [나. 회사의현황의 생산공장개요부분]을 참고 부탁드립니다.
나. 회사의 현황
(1) 회사의 영업 및 생산
당사는 제1공장에 대하여 교차오염에 대한 위험성이 낮고, 설비 단순화 및 신규제품 및 기술에 관한 유연성확보가 가능한 전공정 Single-Use 생산방식의 생산설비를 구축하여, 본 배양 기준 2,000 L Single Use 배양기 3기를 포함한 총 6,000L 규모의 연간 최대 50배치의 생산이 가능한 시설을 갖추었으며, 투입량 기준으로 연간 150,000L의 생산 Capacity를 확보하였습니다.
의약품 시장은 전통적으로 국민의 건강과 복지에 직결되는 산업임에 따라 의약품의 허가 및 시판을 위해서는 각 국가의 규제기관 및 국제기구가 정한 비교적 까다로운 기준인 GMP(Good Manufacturing Practice) 가이드라인에 맞게 진행되어야 합니다. GMP(Good Manufacturing Practice)는 우수한 의약품을 제조하기 위하여 공장에서 원료의 구입부터 제조, 출하 등에 이르는 모든 과정에 필요한 관리기준이며, 당사는 해당 관리기준을 충실히 갖추기 위하여, 최첨단 공정 시스템인 스마트 팩토리 플랫폼을 도입하였습니다. 바이오의약품 제조공장으로서 주요 산업연혁은 다음과 같습니다.
|
일자 |
주요 연혁 |
|
2016.11 |
충북 경제자유구역청과 항체의약품 제조 및 연구시설 설립 지원 MOU 체결 |
|
2017.05 |
충북 오송첨단의료복합단지 제1연구소 및 제1생산공장(6,000리터) 착공 |
|
2017.08 |
충북 오송첨단의료복합단지 제1연구소 및 제1생산공장 착공, 벤처기업 인증 획득(기술평가보증기업), 기업부설연구소 인증 획득 |
|
2017.12 |
충북 바이오폴리스지구 입주 및 매입계약 체결(제2공장 예정지) |
|
2018.02 |
써모피셔사이언티픽사와 SmartFactory™(스마트 팩토리) 플랫폼 구축 MOU 체결 |
|
2018.04 |
중소기업 인증 획득 |
|
2018.06 |
충북 오송 제1연구소 및 제1생산공장 준공 |
|
2018.06 |
본사 이전, 제조업 허가 획득 |
|
2018.11 |
EU QP(Qualified person) Audit 완료 |
|
2019.01 |
오송첨단의료복합단지 제1생산공장 등록 |
|
2019.04 |
중소기업 인증 획득, 의약품 제조 및 품질관리기준(GMP) 적합판정 획득 |
|
2019.08 |
벤처기업 인증 획득(연구개발기업) |
|
2019.11 |
EU GMP 사찰 준비 용 갭 분석 컨설팅 (Gap Analysis Consulting for EU GMP Inspection) |
|
2019.12 |
EU GMP 사찰 준비용 갭 개선 컨설팅 (Remediation Consulting for EU GMP Inspection) |
|
2020.02 |
EU GMP 실사 1차 진행(주) |
| 2021.01 | 프레스티지바이오파마(싱가포르)와 PBP1510 생산계약 체결 |
| 2021.02 | 충북 오송바이오폴리스지구 제2생산공장 착공 |
| 2021.03 | 기업공개 코스닥 상장 |
| 2021.05 | 충북 오송 제1캠퍼스 증축 R&D 센터 착공 |
| 2021.11 | 충북 오송 제1캠퍼스 증축 R&D 센터 준공 |
(주) 2020년 1월 27일부터 EMA 실사를 진행하였으나, 실사 도중 코로나바이러스 감염증-19(COVID-19)으로 인하여 EMA 측의 연기결정으로 중단되어 심사가 지연되고 있습니다. 자세한 사항은 '나.회사의현황 -(1) 회사의 영업 및 생산 - (나)생산공장 개요' 를 참고 부탁드립니다.
당사는 현재 항체의약품 생산 전문제약회사로서 항체의 정제와 관련하여 Protein A를 이용하지 않는 독자적인 항체 정제방법 기술을 보유하고 있으며, 이와 더불어 당 함량 조절을 통한 항체의 제조기술, 이성질체 함량 조절을 통한 항체의 제조기술을 통해 고품질의 항체의약품을 높은 가격 경쟁력으로 생산하고 있습니다. 또한, 현재까지 제1공장을 설계, 운영하며 당사가 축적한 노하우와 기술을 최대로 활용하여 앞으로 만들 제2공장에는 국내 최초로 최첨단 공정 시스템인 Smart Factory(스마트 팩토리) 플랫폼과 함께, 일반적으로 바이오생산 공정에서 개별적으로 사용되는 Stainless Steel 생산방식과 Single Use 생산방식이 결합된 ALITA 플랫폼을 설계 도입할 예정인데 이를 통해, 다양한 제품을 생산하여야 하는 CMO의 사업목적에 부합하는 당사만의 플랫폼 기술을 갖출 계획입니다.
당사는 관계사 프레스티지바이오파마사와의 라이선스계약을 통하여 HD201(허셉틴 바이오시밀러) 및 HD204(아바스틴 바이오시밀러)의 개발에 참여하고 있으며, 전체 개발 과정 중에서 공정 개발 및 품질 관련 분야를 담당하고 있습니다. 이로 인하여 해당 파이프라인에 대하여 일정수익 분배지분율을 보유하고 있으며, 우선제조권한을 보유하고 있습니다. 글로벌제약사와의 기술이전 및 라이선스계약과 공급계약도 함께 체결함으로써 향후 5~ 10년간의 장기계약으로 안정적인 사업환경을 갖추고 있습니다.
(가) 생산활동 개요
바이오의약품의 생산 공정은 일반적으로 「원제공정(배양공정 → 정제공정) → 완제공정」으로 구성되어 있으며, 제조하고자 하는 바이오의약품의 특성에 따라서 세부 공정에서 차이가 있습니다.
|
단 계 |
내 용 |
|
원제공정 (배양공정,Upstream Process) |
배양공정은 기본적으로 CHO Cell의 성장을 위한 조건을제공하고 얻고자 하는 생물학적 활성 성분을 생산하기 위하여 개발 설계되며, 크게 ‘세포주 해동 단계‘, 플라스크 및 증식용 배양기가 사용되는 ‘종배양(Seed Culture) 단계‘, 대용량 생산용 배양기가 사용되는 ‘본배양(Main Culture) 단계‘로 구성됩니다. 이후, 원심분리, 여과 등의 방법을 이용하여 배양액을 회수합니다. |
|
원제공정 (정제공정, Downstream Process) |
정제공정은 최종 회수 이후 첫 단계부터 시작되며 얻고자 하는 품질의 제품 생산까지 이어집니다. 농축 및 불순물 제거를 통해 정제공정이 수행되며, 가장 흔하게는 다양한 크로마토그래피와 여과 방법이 사용됩니다. |
|
완제공정 |
완제공정은 최종완제품을 생산하는 단계로 원액 주성분, 첨가제 등을 혼합하고 무균여과를 실시하는 과정으로 환자가 투여할수 있도록 최종제품으로 충전하고 포장하는 과정입니다. |
다만, 당사는 원료의약품 생산공정(원제공정)만 보유하고 있으며, 완제의약품 생산공정의 경우 원료의약품 생산공정에 비교하여 기술의 복잡성이 낮으며, 설비구축 비용 및 경제적 효익 등을 종합적으로 고려하여 자체 생산보다는 위탁 생산하는 것이 효율적인 것으로 판단되어 완제의약품 생산공정은 국내외에 소재하는 각국의 GMP를 획득한 회사에 위탁하여 생산하고 있습니다.
(나) 생산공장 개요
당사의 본사와 제1공장은 충북 오송에 위치하고 있습니다. 2018년 6월 충북 오송첨단의료복합단지에 총 생산 Capacity가 6,000L인 제1생산공장(연면적: 10,949.71㎡)을 준공하였으며, 이후 7개월만인 2019년 1월, 주요 생산장비 및 생산지원설비의 설치·운전 적격성 평가부터 우수의약품 제조관리기준(Good Manufacturing Practice, GMP) 인증 획득을 위한 공정 밸리데이션 (Process Validation) 배치 생산 및 실사를 완료하여 2019년 4월 한국 식품의약품안전처로부터 의약품 제조 및 품질관리기준 적합판정 획득하였습니다. 또한, 관계사의 글로벌제약사와의 라이선스 및 제품공급 계약 이전에 수차례 생산 현장 실사를 진행하여 Quality 안정성을 증명함으로써 실제 계약까지 달성하고 있습니다.
당사는 글로벌 CDMO 시장에서의 경쟁력을 갖추기 위하여 98,000L 생산규모의 제2공장 설립하여, 제조 라인을 다양화하여 안정적인 생산환경 및 효율적인 생산시스템을 구축할 계획입니다. 또한, 공정개발과정에서 원가를 절감할 수 있는 다양한 특허를 보유하고 있으며, 더불어 자체적인 엔지니어링 기술을 보유하고 있어 최적화된 공정을 구현하여 효율적인 생산 서비스를 제공하고 있습니다.
|
구분 |
제1공장 |
제2공장 |
R&D센터 |
|
위치 |
충북 오송 첨단의료복합단지 (오송읍 연제리 654) |
충북오송 바이오폴리스 지구 (오송읍 정중리799) |
충북오송 첨단의료복합단지 (오송읍 연제리 654-1) |
|
대지면적 |
10,560 ㎡ |
25,000 ㎡ |
6,681㎡ |
|
연면적 |
10,949.71㎡ |
32,900 ㎡ |
15,352.82 ㎡ |
|
규모 |
6,000 L (2,000L X 3기) |
98,000 L (1차: 2,000L X 14기, 2차: 5,000L X 14기) |
24,800 L 8,000L (2,000L X 4기) 800L (200L X 4기) 16,000 (2,000 X 8기) |
|
공사기간(착공-준공) |
2017.08-2018.06 |
1차: 2021.02-2021.12 2차: 2022.01-2022.12 |
2021.03-2021.11 |
(주) 신규시설투자에 대한 자세한 사항은 2021년 4월19 일(신규시설투자-제2공장) 및 2021년 6월 10일(신규시설투자-R&D센터 외) 공시사항을 참조하여 주시기 바랍니다.
당사의 제1공장은 HD201(허셉틴 바이오시밀러)의 유럽지역의 품목허가를 위하여 다음과 같이 EU-QP(Qualified Person)로부터 EU-GMP 예비사찰을 성공적으로 마쳤습니다.
|
규제기관 |
실사 일자 |
현황 |
|
EU QP Inspection (유럽 QP) |
2018년 10월 29일 ~ 2018년 11월 2일 |
Approved(승인) |
이 후 2019년 4월 EMA 판매허가 신청에 따른 EU-GMP 실사를 완벽히 대응하기 위하여, 추가적으로 하기와 같이 전문 컨설턴트로부터 실사를 진행하여 제1공장의 EU-GMP 인증을 위한 준비를 모두 마쳤습니다.
|
자문 회사 및 컨설턴트 |
자문 유형 |
자문 일자 |
비고 |
|
PQE, Italy |
Initial/Gap Assessment |
2019년12월 05일~07일 |
EU GMP 적합 평가 |
|
교정 자문 (Remediation) |
2019년12월 16일~21일 | ||
|
사전 실사 (Mock Inspection) |
2020년01월 16일~18일 |
다만, 2020년 1월 27일부터 EMA 실사를 진행하였으나, 실사 도중 코로나바이러스 감염증-19(COVID-19)으로 인하여 EMA 측의 연기결정으로 중단되어 심사가 지연되고 있으며 자세한 내용은 다음과 같습니다.
▣ 제1공장 EU - GMP실사관련
2020년 1월 27일부터 EU-GMP실사를 진행하였으나, 실사 도중 코로나바이러스 감염증-19(COVID-19)의 전세계적 확산으로 인한 EMA 측의 연기결정으로 실사가 연기되었습니다. 2020년 8월 유럽 EMA는 관계사 프레스티지바이오파마의 HD201에 대하여 유럽 EMA품목 허가 일정 재개를 권고하였고, 이에 생산시설에 대한 EU-GMP 실사여부와 관계없이 품목허가 심사를 재개하던 중 2021년 3월 22일 심사단계인 D210에서 품목허가는 EU-GMP 현장실사를 실시하는 것으로 결정이 되었습니다. 하지만 코로나 19의 지속된 확산으로 인해 EU-GMP 실사단의 방문 일정이 확정되지 않았습니다. 이에 당사는 조속한 EU-GMP 현장실사를 위해 지속적인 노력을 하고 있습니다. EU-GMP의 현장실사 이후 승인권고에 대한 일정은 순차적으로 진행 될 예정입니다.
(다) 생산제품 개요
당사는 싱가포르에 소재하는 관계사인 프레스티지바이오파마(Prestige Biopharma Limited)의 파이프라인인 HD201(허셉틴 바이오시밀러) 및 HD204(아바스틴 바이오시밀러)에 대해 2015년 라이선스계약을 체결하였습니다. 전체 개발과정 중 공정 개발 및 임상의약품생산, 상업화 공정 등을 위한 연구를 진행하며 일정개발비용을 부담하고 두 파이프라인에 대한 우선제조권한 및 해당 파이프라인의 라이선스아웃(License-out)계약체결을 통해 창출되는 수익(로열티 등)에 대한 일정 %의 수익분배지분율을 확보하여 향후 5~10년간의 안정적인 성장기반을 마련하였습니다.
현재 EMA(유럽) 허가기관으로부터 판매승인 심사가 진행하고 있는 HD201(허셉틴 바이오시밀러)의 경우 2022년 상업화 생산을 앞두고 있으며, 글로벌 임상 3상이 진행 중인 HD204(아바스틴 바이오시밀러)의 경우 임상의약품 생산, 공정개발 연구 및 CPO서비스를 제공하고 있습니다. 이 외에도, 현재 신약으로서의 성공가능성을 인정받아, FDA(미국), MFDS(한국) 및 EMA(유럽) 허가기관으로부터 희귀의약품 지정을 승인받은 프레스티지바이오파마의 췌장암 신약파이프라인(PBP1510)에 대한 임상의약품 제조 및 공정개발업무도 진행하고 있습니다.
|
파이프라인 |
계약 내용 |
비고 |
|
HD201 |
공동개발 및 제조 |
제품판매매출(주1) 로열티에 대한 수익일정지분 권리 보유(Profit sharing) |
|
HD204 |
공동개발 및 제조 |
제품판매매출(주1) 로열티에 대한 수익일정지분 권리 보유(Profit sharing) |
|
PBP1510 |
위탁개발 및 위탁제조 |
위탁개발 및 임상의약품 위탁제조(주2) |
(주1) HD201 및 HD204에 대한 공급계약은 프레스티지바이오로직스가 우선제조권 및공급권한에 따라 공급계약의 실질적인 주체이며, 프레스티지바이오파마는 공급계약체결권을 보유하고 있습니다. 이에 따라 공급계약 이행에 따른 수익 및 책임은 모두 당사에게 귀속됩니다.
(주2) 위탁사인 프레스티지바이오파마사의 파이프라인 PBP1510의 임상 1상용 의약품 제조를 위한 배양, 정제 공정의 개발 및 최적화, 다양한 분석시험법 확립 및 제품 품질특성 분석, 제조소에서의 생산을 위한 제조공정 기술 이전, 그리고 GMP 생산, 출하시험, 최종 완제의약품 생산 및 안정성 시험까지 포함된 CDMO 계약.
당사가 개발 및 생산을 진행하고 있는 제품에 대한 주요 내용은 하기와 같습니다.
▣ HD201(허셉틴 바이오시밀러)
|
구분 |
내용 |
|
① 개발사 |
프레스티지바이오파마(Prestige Biopharma Limited) |
|
② 구분 |
바이오시밀러 |
|
③ 적응증 |
유방암, 전이성 위암 등 |
|
④ 성분명 |
트라스트주맙 (trastuzumab) |
|
⑤ 작용기전 |
트라스트주맙은 HER2를 표적하는 단일클론 항체치료제이며, HER2 수용체가 과발현되어 있는암세포에 효과적으로 작용하고 주로 유방암 치료제로 사용됨. 이들은 HER2 수용체와 결합하여 과발현된 HER2 신호전달경로를 차단함으로써 암의 성장, 발달을 억제하며, 또한 항체가 갖는 불변 부위(Fc)에 의해 일어나는 항체의존 세포독성 (ADCC; antibody-dependent cell-mediated cytotoxicity)에 의해 암세포의 사멸 효과를 얻을 수 있음 |
|
⑥ 제품의 특성 |
총 502명의 HER2 양성 유방암 환자를 대상으로 전 세계 13개국에서 진행된 임상 3상 시험 결과, 유방 및 액와부(겨드랑이) 림프절 종양이 완전히 소실된 환자의 비율(병리학적 완전관해율)과 유방조직 내 종양이 완전히 소실된 환자의 비율(유방조직 완전관해율) 평가에서 기존 표적치료제인 허셉틴과 각각 0.5%, 1.7% 이내의 반응 차이를 확인함. 글로벌 임상 3상을 통하여 안전성 및 원약과의 비교동등성 99.5% 입증하면서 현재까지 발표된 허셉틴 바이오시밀러 중 가장 높은 유사성을 보임. |
|
⑦ 진행경과 |
- 글로벌 임상 3상 개시: 2018년 1분기 - Bridging Study 개시: 2019년 1분기 - 글로벌 임상 3상 결과보고서(iCSR) 완료: 2019년 2분기 - 유럽 품목허가 신청 완료: 2019년 2분기 - 미국 FDA 허가 신청 사전미팅: 2019년 3분기 - 캐나다 보건국 허가 신청 사전미팅: 2020년 1분기 - Bridging Study 완료: 2020년1분기 - 유럽EMA 품목허가 승인 심사 중 |
|
⑧ 향후계획 |
- 미국 FDA 품목허가 신청: 2021년 (목표) - 캐나다 보건국 품목허가 신청: 2021년 (목표) - 한국 MFDS 품목허가 신청: 2021년 (목표) |
|
⑨ 경쟁제품 |
- Ogivri, Ontruzant, Herzuma, Kanjinti 등 |
|
⑩ 관련논문 등 |
*유럽 종양학회 정기 학술대회(ESMO) 2020: 임상 연구결과 발표 [학회 발표] A phase III trial to compare the efficacy, safety, pharmacokinetics and immunogenicity of HD201 to trastuzumab in HER2+ early breast cancer patients (TROIKA) [학회 발표] A double-blind, randomized, parallel group study to demonstrate the equivalent pharmacokinetic properties of a single intravenous dose HD201, a trastuzumab biosimilar candidate, versus EU trastuzumab and US trastuzumab *미국 임상종양학회(ASCO) 2020: 임상 연구결과 발표 [학회 발표] Establishing analytical and clinical similarity between HD201 and herceptin *논문: A Randomized Phase I Study Comparing the Pharmacokinetics of HD201, a Trastuzumab Biosimilar, With European Union-sourced Herceptin, Clinical Therapeutics, Volume 40, Issue 3, March 2018, Pages 396-405.e4 등 다수 논문/학회 발표 진행 |
|
⑪ 시장규모 |
오리지널 의약품 글로벌 시장규모: 2019년 기준 $ 6,078 million (출처: GlobalData) |
|
⑫ 기타사항 |
[라이선스계약]2015년 7월에 최초 체결된 라이선스계약을 토대로 당사는 HD201개발과정 중 제조와 관련된 공정개발 및 품질관련분야를 담당 [라이선스아웃 및 판매계약] II.사업의내용-9.경영상의 주요계약 등 참고 |
▣ HD204(아바스틴 바이오시밀러)
|
구분 |
내용 |
|
① 개발사 |
프레스티지바이오파마(Prestige Biopharma Limited) |
|
② 구분 |
바이오시밀러 |
|
③ 적응증 |
전이성 직결장암, 비소세포폐암, 진행성 또는 전이성 신세포암, 교모세포종, 상피성 난소암, 난관암 또는 원발성 복막암, 자궁경부암 |
|
④ 성분명 |
베바시주맙 (Bevacizumab) |
|
⑤ 작용기전 |
베바시주맙은 수용성VEGF에 결합하여VEGF가VEGFR과 상호작용하는 것을 억제하는 인간화 단일클론 항체임. VEGF에 대한 항체의 결합은VEGF가 그것의 수용체와 결합하는 것을 방해하여VEGF에 의한 생물학적 활성 억제를 유도함. VEGFR은 정상 조직 혈관에 비해 종양의 맥관구조(Vasculature) 내피(Endothelium)에 과발현 되어있기 때문에 베바시주맙은VEGF와 결합함으로써 종양의VEGFR 활성을 억제하여 새로운 혈관형성 활성을 억제함. 혈관 형성을 억제되면 종양 세포에 대한 산소와 영양분 공급이 어려워지고, 노폐물이 축적되어 종양 세포가 생존하기 어려운 환경이 조성되어 종양 성장을 억제함. |
|
⑥ 제품의 특성 |
호주에서 진행된 임상 1상 결과, 약동학(PK) 측면 및 안전성 모두에서 US-Avastin 및 EU-Avastin과의 동등성이 우수함이 입증되었음 |
|
⑦ 진행경과 |
- 유럽 임상 1상 개시: 2018년 3분기 - 유럽 임상 1상 완료: 2019년 2분기 - 글로벌 임상3상 개시: 2019년 2분기 - 글로벌 임상3상 진행 중 |
|
⑧ 향후계획 |
- 미국FDA 품목허가 신청: 2021년(목표) |
|
⑨ 경쟁제품 |
Mvasi, Zirabev, Krabeva 등 |
|
⑩ 관련논문 등 |
*미국 임상종양학회(ASCO) 2020: 임상 연구결과 발표 [학회 발표] Assessment of quality and clinical similarity (pharmacokinetic and safety) of HD204, a biosimilar of bevacizumab |
|
⑪ 시장규모 |
오리지널 의약품 글로벌 시장규모: 2019년 기준 $ 7,118 million (출처: GlobalData) |
|
⑫ 기타사항 |
[라이선스계약]2015년 7월에 최초 체결된 라이선스계약을 토대로 당사는 HD204개발과정 중 제조와 관련된 공정개발 및 품질관련분야를 담당 [라이선스아웃 및 판매계약] II.사업의내용-9.경영상의 주요계약 등 참고 |
▣ PBP1510(췌장암 항체신약)
| ①구분 | 항체신약 |
| ②적응증 | 췌장암, 난소암 |
| ③작용기전 | 인체 내에 존재하는 췌장암 전이인자 PAUF를 중화시켜 자가분비 및 주변분비 신호 전달을 감소 |
| ④제품의 특성 |
췌장암 치료 항체신약으로서의 성공 가능성을 인정받아 미국 FDA, 한국 MFDS, 유럽 EMA로부터 희귀의약품 지정을 승인받음. PBP1510은 희귀의약품으로 지정됨에 따라 임상 2상 임상시험 자료만으로도 판매 허가할 수 있는 조건부 허가와 우선 심사 및 신속 절차, 임상 비용 지원, 신약 시판 승인을 위한 시험 계획 자문, 그리고 시장 독점 등 다양한 혜택의 기회를 얻음 |
| ⑤진행경과 |
- Chemical Abstracts Service (CAS) 등록: 2020년 2분기 - WHO 국제일반명(INN) 신청: 2020년 2분기 - 희귀의약품지정 미국FDA 승인: 2020년 2분기 - 희귀의약품지정 한국MFDS 승인: 2020년 3분기 - 희귀의약품지정 유럽EMA 승인: 2020년 4분기 - 유럽 및 국내 임상 1/2a상 준비 중 |
| ⑥향후 계획 |
- 유럽 및 국내 임상 1/2a상 개시: 2021년 4분기 (예정) - 유럽EMA 및 미국FDA 품목허가 신청: 2024년1분기(목표) |
| ⑦경쟁제품 | N/A |
| ⑧관련논문 등 | [특허] PAUF 단백질에 특이적으로 결합하는 항체 및 이의 용도 |
| ⑨시장규모 | N/A |
(라) 기술경쟁력 개요
당사는 HD201, HD204의 상업화업무를 진행함에 있어 의약품생산만을 담당한 것이 아닌 당사의 특허기술 확장을 비롯하여 해당 파이프라인의 공정개발 및 대체원료 개발을 통해 원가경쟁력을 확보하기 위한 개발업무를 다년간 진행하였습니다.
① 특허생산기술
제1공장의 전공정 Single-use 시스템 채택, Upstream(배양) 및 Downstream(정제) 공정의 완벽한 분리로 오염에 대한 리스크 및 이로 인한 실패위험을 감소시키고, 생산 공정 중 배양에서는 N-1 Perfusion (Cell 농축)의 적용으로 생산기간 단축 및 배양기 숫자를 절감하여 효율적이고 경제적인 공정을 구현하였습니다. 특히, 중공사막필터(Hollow Fiber Filter, HF)를 이용한 TFF (Tangential Flow Filtration)를 Perfusion 공정에 적용함으로써 일반적으로 사용되는 ATF (Alternating Tangential Flow Filtration)에 비해 비용 면에서 한층 더 유리한 공정 구축하였습니다. 정제공정에서는 단일클론 항체 정제에 일반적으로 사용하는 Protein A 크로마토그래피(Lock and Key 원리로 정제 효율이 높으나 고가의 레진 사용)가 아닌 양이온 교환 크로마토그래피를 사용하여 정제하는 기술을 확보, 적용함으로써 Protein A 정제에서 얻을 수 있는 동등한 효율로 항체의 품질은 유지하면서 비용은 획기적으로 절감시킬 수 있는 공정을 적용하여 보다 가격 경쟁력 있는 생산이 가능하도록 하였습니다. 해당 정제방법에 대한 특허정보진흥센터는 특허 생존지수 295.1(MAX 300)은 상위 5%에 포함되는 S등급으로 평가되며, 매우 우수한 특허로 분류되었습니다. 상업성면에서도, 유사기술군 평균 64.7%에 비해 월등히 높은 84.0%를 보여주었으며, 2032~2033년까지 한국, 미국, 일본, 유럽, 중국에서 독점적 유지가 가능하고, 유사한 성질을 가진 항체(단백질)정제에서도 활용하는 등 유사기술의 확장 가능성이 있다고 분석되고 있습니다(특허정보진흥센터 분석, 2019.08.30).
② 대체원료 개발
항체 의약품의 생산 공정에서 배양 공정을 통하여 세포를 증식시키며, 증식에 따른 단백질 발현 정도에 의해 생산성이 결정됩니다. 또한, 단백질을 발현하는 세포는 살아있는 세포로서 이의 증식과정에서 영양분을 공급하는 배양액과 세포주의 적합성이 생산성에 많은 영향을 주고 있습니다. 당사는 세포주에 따른 각각의 다른 영양 요구 특성으로 인해 일반적으로 시장에서 구입할 수 있는 배양액으로는 각각의 특성에 따른 다른 영양 요구를 반영할 수 없다고 판단하여, 각 파이프라인별 대체원료개발을 진행해 왔으며, 그 중 2017년부터 국내의 협력사와 HD201(허셉틴 바이오시밀러)에 적합한 배양액의 공동 개발을 진행한 결과 임상 1, 3상 시료 생산 시 사용되었던 13 종의 배양 첨가물들을 단일 제품화하여, 세포 배양 배지 준비 공정의 효율성을 증대시키고 항체 발현에 필수적으로 요구되는 L-methionine sulfoximine(MSX)의 원료의 단가를 획기적으로 감소시켰습니다.
이후, HD201 항체의 생산성 증대를 위하여, 세포 배양액 교체 연구를 수행하였으며, 현재 연구용 규모의 배양에서 약 2배 정도의 생산성을 확보하였습니다. 기존 공정의 규제기관(유럽 EMA) 승인 이후, 신규 공정에 대한 허가 획득을 위해 설계기반 품질고도화 (QbD) 연구를 수행 중이며 이를 통해 기존 공정과 동등한 품질이 확보된 공정을 확립할 예정입니다.
PBP1502의 경우, 2019년 200L scale에서 수 g/L 이상의 항체 발현률을 보이는 공정을 확립하였습니다. 현재 동사에서는 기존 공정보다 30% 이상 생산성이 높으며 동시에 세포 배양 배지 단가를 낮출 수 있는 대체 배양액을 협력사와 공동 개발 중이며 그 결과 기존대비 약 50%정도 이상의 항체 발현할 수 있는 대체 배양액을 확보하였습니다
[대체원료 개발 진행과정]
|
구분 |
내용 |
|
2017년 03월 |
GS supplement Premixture Project 개시 |
|
2018년 04월 |
HD204 배양액 배지 개발 착수 |
|
2018년 03월 |
PBP1502/1510 배양 배지 개발 착수 |
|
2018년 06월 |
PBP1502/1510 초기 배양 배지 선정 |
|
2018년 07월 |
GS supplement Premixture 시제품 개발 완료 |
|
2018년 08월 |
PBP1502/1510 Basal Media/Feed 배지 선정 완료 |
|
2018년 09월 |
HD201 GS supplement Premixture 2000L 생산 투입 |
|
2018년 09월 |
HD201 신공정 파일럿 배지 출시 완료, Test 배양 실시 |
|
2018년 11월 |
PBP1502/1510 Basal Media/Feed 배지 screening 완료 |
|
2019년 08월 |
PBP1502, 고발현하는 세포 배양액 확보 완료 |
|
2020년 03월 |
HD201 신공정 배지 결과 확보 |
③ 최적화된 생산공정 구축기술
오송 제1공장은 설립당시 글로벌임상 진행중인 HD201(허셉틴 바이오시밀러), HD204의 임상의약품 공급 및 생산효율을 최대한으로 높일 수 있도록 공정을 설계하였으며, 공정 변경이 용이하여 유연성이 높은 Single Use 생산방식을 채택하여 고가의 턴오버(Turn-over) 비용을 최소화할 수 있도록 독창적인 설계 및 구현에 성공하였습니다.
이후, 2021년 상반기부터 본격적인 상업 생산에 따른 대량생산 및 급부상하고 있는 바이오의약품 위탁서비스 수요에 대응하기 위하여 제1공장은 다품종ㆍ소규모생산, 제2공장은 다품종ㆍ대규모생산에 가장 최적화된 생산공정을 구축하여 고객사의 가격 경쟁력 향상을 지원하게 될 것입니다.
▣ 전공정-Single Use 생산방식 국내 최초 도입
바이오의약품은 효능의 발전만큼이나 제조방식에서도 많은 발전이 있었으며, 바이오의약품 제조공정에서 Cross Contamination(교차감염)이 중요한 리스크로 부상하게 되었습니다. 특히, 현재 경쟁사가 채택하고 있는 Stainless Steel 배양 방식에서는 대용량 생산이 가능한 장점이 있는 반면에, 오염에 노출 될 경우, 근본 원인의 파악 및 정상가동화에 많은 노력과 시간을 투자해야 하는 단점이 있습니다.
교차오염을 예방하기 위하여 1회용 Bag을 활용한 Single use 생산 방식이 새롭게 등장하였고, 당사는 교차오염을 최소화하고자 국내에서 최초로 전공정 Single use에 따른 생산 방식을 채택하고 있습니다. Single use(1회용 Bag 사용) 방식은 교차오염 Risk가 매우 적고, 감염이 된 경우라 하더라도 해당 소모품만을 교체함으로써 Validation 재확인 등 재가동 공백기간이 수일 이내 이므로 경제성이 높습니다. 단, 1회용 Bag 등 소모품 비용이 상대적으로 비싼 단점이 있으나, Single use 방식이 점차 보편화됨에 따라 소모품의 부품 소재화 사업이 확대되면서 소모품의 조달 비용도 낮아지고 있는 추세입니다.
|
방식 |
Stainless Steel |
Single Use |
|
장점 |
- 대용량/batch 생산 가능 - Single use방식 대비 소모품비 낮음 |
- Cross Contamination Risk 없음 - 신속한 배치전환 가능(동일한 규모의 Stainless Steel 방식에 비교하여 생산소요 기간이 약 60% 정도 짧음) - 초기 투자비 낮음 - 설비 유지비용 낮음(Cleaning 없음) |
|
단점 |
- 초기 투자비 높음 - 반복 사용으로 인한 Cross Contamination Risk - 설비 유지비용 높음(주기적 Cleaning 작업필요) - 배치 전환 준비 소요시간 |
- 생산량 한계: Max 2,000L (상용화 기준, 일부제조사에서 2,000L 이상도 제조함) - Stainless Steel 방식 대비 소모품비의 비중이 높음 |
▣ ALITA의 배치증산
당사는 제1공장의 엔지니어링 기술과 전공정 Single Use 시스템의 경험을 바탕으로 고객에게 최소한의 비용으로 제조공정에 따라 최적화된 장비 등 시스템을 제공하고 Single Use 대비 낮은 비용적 부담을 발생시키는 생산방식을 고안하였으며 이는 Stainless Steel 생산방식과 Single Use의 장점을 결합한 당사만의 특화된 생산 방식을 제2공장부터 도입할 계획이며, 이를 ALITA Smart Bio Factory 또는 ALITA로 명명하였습니다. ALITA는 높은 유연성 확보와 빠른 배치회전 확보에 기술적 주안점(주1)을 두었으며, Single Use System과 비교시 다음과 같은 장단점을 지니고 있습니다.
주1) 현재 이와 관련된 특허가 진행 중으로 대외비사항은 기재하지 않았습니다.
|
구분 |
Single-use |
ALITA |
|
장점 |
낮은 투자 및 운영 비용 오염 리스크 낮음 제품전환 및 공정변경 용이 |
빠른 회전율 제품전환 및 공정변경 용이 낮은 소모품 비용 자동화 구현 |
|
단점 |
Scale Limit (Bioreactor : 2000L ~ 5000L, Mixer : 5000L) 1회용 소모품 비용 높음 작업자 의존성 높음 |
소량생산시 장점활용도 저하 SS와 SU 연결에 무균 접합기술(동사 특허출원 기술) 필요 |
(2) 시장점유율
현재 당사는 CMO 및 CDO부문의 매출액이 없어 매출액 기준으로 시장점유율을 판단하기에는 다소 어려움이 있습니다. 일반적으로 바이오의약품 생산 규모는 CMO의 매출에 많은 영향을 주며, 현재 36.4만리터의 배양설비를 보유하고 있는 삼성바이오로직스, 독일과 미국 등에 31.9만리터 생산설비를 보유하고 있는 독일의 Boehringer Ingelheim, 29만리터의 배양설비를 보유하고 있는 스위스의 Lonza가 CMO 시장에서 선두 CMO그룹을 형성하고 있으며, 그 외 국외의 Asahi Glass(36,000L), Thermo Fisher(18,500L) 를 비롯하여 국내의 바이넥스(11,200L)등이 시장을 형성하고 있습니다.
당사는 현재 제1공장은 6,000L 규모로 운영되고 있으며, 2022년 가동예정인 제2공장 Phase1는 28,000L 규모로 2021년 2월 착공하여 2022년 12월 준공을 목표로 하고 있으며, Phase2는 70,000L 규모로 2022년 착공하여 2023년 가동될 예정입니다.
(3) 신규사업 등 내용전망
당사는 임상의약품의 위탁개발 및 위탁제조 뿐 아니라 맹검포장, 보관 및 운송까지의 풀서비스를 제공하는 CPO(Contract Packaging Organization)사업 및 최단기내 고객이 원하는 생산이 가능한 맞춤식 제조시설 구축을 위한 Suite Engineering 서비스를 제공하는 CEO(Contract Engineering Organization)사업까지 서비스 범위를 확대하고 있습니다.
[CPO]
바이오의약품의 수요증가로 신약에 대한 연구 및 개발도 성장하는 가운데, 임상시험에서 사용되는 임상의약품 포장에 필요한 특화된 서비스를 제공하는 전문 국내기업이 부족하여, 국외기업에 의존하고 있는 것이 현재의 실정입니다. 이에 당사는 개발사가 최종 임상 대상 환자에게 적절하고 통제된 임상의약품을 공급하게 하기 위하여, 포장, 보관 및 운송을 포함하는 임상 포장 서비스를 제공하고 있습니다. 여기에는 라벨 제작, 전문적인 품질 문서 관리, 라벨링, 디라벨링 및 온도 조절 운송ㆍ보관이 포함됩니다.
|
서비스 내용 |
내 용 |
|
Label Management |
- 단일 라벨 및 다중 라벨 제작 - 각 국가에 맞는 언어 번역 및 규제 지원 - 통제된 라벨 관리 시스템 |
|
Labeling & Documentation |
- 임상약 특징에 맞는 라벨링 시스템 도입 - 각 고객의 요청에 따른 문서 제작 및 관리 - GMP 수준에 따르는 임상포장실 |
|
Transportation & Storage |
- 고객의 요청에 부합하는 운송 및 보관 서비스 - 실시간 관리되는 냉장보관 서비스 |
해당 사업은 바이오의약품시장의 성장과 함께 성장하는 사업입니다. 현재 CPO사업의 경우, 글로벌 임상3상을 진행하고 있는 HD204(아바스틴 바이오시밀러) 파이프라인을 대상으로 서비스를 제공하고 있으며, 물적자원보다 인적자원이 필요한 사업으로 현재 관련장비는 모두 갖추어 향후 자금의 투입은 크지 않을 것으로 예상되고 있습니다. 또한 현재 글로벌 기준의 품질관리 및 유지를 통해 CPO서비스를 제공하기 위하여, WHO기준에 따른 GDP(Good Distribution Pratice, 우수유통관리기준)인증을 위한 실사를 완료하였으며 2021년 4월 인증을 획득하였습니다.
[CEO]
바이오벤처기업의 경우 공정개발과 시설구축에 요구되는 전문인력의 확보, 투자비용, 허가 및 시장진입 등 제한조건을 가지고 있어 자체적으로 상용화 생산단계까지 제품을 개발하는 것이 불가능하며, 기존 제조설비를 보유한 글로벌제약사의 경우에도 생산성 제고를 위한 엔지니어링적 개선에 한계가 있으며, 신규 제조시설의 구축에는 막대한 비용 및 시간이 소요되는 현실입니다.
기존 CMO 및 CDMO의 경우 의약품의 위탁생산과 연구개발에 일부 참여하는 서비스를 제공하고 있기는 하나 엔지니어링 측면에서는 한계를 가지고 있습니다. 위탁생산을 수행하는 업체는 기존에 보유하고 있는 생산 설비와 제조공정을 이용하여 새로운 종류의 제품을 주문받은 양만큼 생산합니다. 기존 공정을 그대로 사용하는 방식으로는 고객사의 제품생산에 최적화된 제조라인을 구현하는 것이 불가능하므로 새로운 생산제품에 맞추어 제조공정을 수정하는 절차를 거쳐야 합니다. 또한 보유한 공정을 일부 수정한다고 하더라도 고객사의 제품 생산성 및 공정효율성을 달성하지 못할 가능성도 있습니다.
이에 당사는 기존 CMO, CDMO사업의 이러한 한계를 극복하고, 차별적인 서비스를 제공하기 위하여 당사가 고안한 신개념 Upgrade Technology Service Platform으로 Bio-Manufacturing에 엔지니어링적인 기술을 접목한 사업을 발족하였습니다.
|
서비스 내용 |
설명 |
|
Process optimization engineering |
개발단계에서 확립된 공정을 분석하여 엔지니어링 단계에서 최적 설계수행 |
|
Productivity enhancement & Cost reduction engineering |
공정최적화를 통한 효율적인 장비사양 도출, 병목해소를 통한플랜트 회전율 확보, 제품 변경을 위한 유연성 확보, 투입되는원부자재 비용 기술적 최적화로 합리적 제품비용 확보, |
|
Conceptual & basic engineering (for manufacturing facility) |
상업 생산규모에서 Mass Balance & Process Flow, 생산장비 요구사양, 요구장비 목록, 유틸리티 및 전기사용량 도출, TimeSchedule 분석, 제조소 Layout 업무 수행 |
|
Customized manufacturing suite |
상기 엔지니어링 활동에 따라 제품 생산에 최적화된 설비에서제조가 가능함에 따라 제품의 생산성을 제고하고 투입원가에 대한 절감이 가능, 유연성을 최대화한 제조소의 제공으로 제품전환에 따른 시설변경에 발생하는 비용을 최소화하여변경에 대한 비용 최소화 가능 |
해당 사업은 엔지니어링 부문의 혁신을 통해 전세계 제약회사에서 바이오 벤처기업까지 아우르는 모든 고객을 위한 전용 맞춤형 제조공간의 제공은 물론, 상업생산에 있어 높은 효율성과 비용절감효과를 누릴 수 있는 의약품 생산에 있어 원스톱 솔루션을 제공하는 것을 당사의 신규 사업으로 발전시키고자 합니다. 현재 당사는 해당사업을 위한 특허를 출원하였으며, 제2공장 준공에 따라 본격적인 사업을 진행할 예정입니다.
2. 주주총회 목적사항별 기재사항
□ 정관의 변경
가. 집중투표 배제를 위한 정관의 변경 또는 그 배제된 정관의 변경
| 변경전 내용 | 변경후 내용 | 변경의 목적 |
|---|---|---|
| - | - | - |
나. 그 외의 정관변경에 관한 건
| 변경전 내용 | 변경후 내용 | 변경의 목적 |
|---|---|---|
|
제12조 [신주의 배당기산일] ①본 회사가 정한 배당기준일 전에 유무상증자 및 주식배당에 의하여 신주를 발행한 경우, 발행한 주식에 대하여는 동등배당한다. ②제1항의 규정에도 불구하고 배당기준일을 정하는 이사회 결의일로부터 그 배당기준일까지 발행한 신주에 대하여는 배당을 하지 아니한다. |
제12조 [신주의 배당기산일] ①본 회사가 정한 배당기준일 전에 유무상증자 및 주식배당에 의하여 신주를 발행한 경우, 발행한 주식에 대하여는 동등배당한다. ②(삭제) |
구주와 신주 모두에게 동등하게 이익배당 |
| 제6장 감 사 | 제6장 감사위원회 | 감사위원회 도입으로 인한 조항 변경 |
|
제49조 [감사의 수] 회사는1인 이상3인 이내의 감사를 둘 수 있다. |
제49조 [감사위원회의 구성] ①회사는 감사에 갈음하여 제45조의 규정에 의한 감사위원회를 둔다. ②감사위원회는 3인 이상의 이사로 구성하고, 총 위원의 3분의 2 이상은 사외이사이어야 한다. ③감사위원회위원의 해임은 출석한 주주의 의결권의 3분의 2 이상의 수로 하되, 발행주식총수의 3분의 1 이상의 수로 하여야 한다. ④ 제3항?제4항의 감사위원회위원의 선임 또는 해임에는 의결권 있는 발행주식총수의100분의3을 초과하는 수의 주식을 가진 주주(최대주주인 경우에는 사외이사가 아닌 감사위원회위원을 선임 또는 해임할 때에 그의 특수관계인, 최대주주 또는 그 특수관계인의 계산으로 주식을 보유하는 자, 최대주주 또는 그 특수관계인에게 의결권을 위임한 자가 소유하는 의결권 있는 주식의 수를 합산한다)는 그 초과하는 주식에 관하여 의결권을 행사하지 못한다. |
감사위원회 도입으로 인한 조항 변경 |
|
제50조 [감사의 선임] ①감사는 주주총회에서 선임 또는 해임한다. ②감사의 선임 또는 해임을 위한 의안은 이사의 선임 또는 해임을 위한 의안과는 별도로 상정하여 의결하여야 한다. 이사회의 결의에 관하여 특별한 이해관계가 있는 자는 의결권을 행사하지 못한다. ③감사의 선임은 출석한 주주의 의결권의 과반수로 하되, 발행주식총수의 4분의 1이상의 수로 하여야 한다. 다만, 상법 제368조의4제1항에 따라 전자적 방법으로 의결권을 행사할 수 있도록 한 경우에는 출석한 주주의 의결권의 과반수로써 감사의 선임을 결의할 수 있다. ④감사의 해임은 출석한 주주의 의결권의 3분의 2 이상의 수로 하되, 발행주식총수의 3분의 1 이상의 수로 하여야 한다. ⑤제3항·제4항의 감사의 선임 또는 해임에는 의결권 있는 발행주식총수의100분의3을 초과하는 수의 주식을 가진 주주(최대주주인 경우에는 그의 특수관계인, 최대주주 또는 그 특수관계인의 계산으로 주식을 보유하는 자, 최대주주 또는 그 특수관계인에게 의결권을 위임한 자가 소유하는 의결권 있는 주식의 수를 합산한다)는 그 초과하는 주식에 관하여 의결권을 행사하지 못한다. |
제50조 [감사위원의 분리선임/해임] ①제49조에 따라 구성하는 감사위원회의 감사위원 중 1명은 주주총회 결의로 다른 이사들과 분리하여 감사위원회위원이 되는 이사로 선임하여야 한다. ②제1항에 따라 분리선임한 감사위원회위원을 해임하는 경우 이사와 감사위원회위원의 지위를 모두 상실한다. |
감사위원회 도입으로 인한 조항 변경 |
|
제51조 [감사의 임기와 보선] ①감사의 임기는 취임 후 3년 내의 최종의 결산기에 관한 정기주주총회 종결시까지로 한다. ②감사 중 결원이 생긴 때에는 주주총회에서 이를 선임한다. 그러나 정관 제49조에서 정하는 원수를 결하지 아니하고 업무수행상 지장이 없는 경우에는 그러하지 아니한다. |
제51조 [감사위원회 대표의 선임] 감사위원회는 그 결의로 위원회의 대표를 선임하여야 한다. |
감사위원회 도입으로 인한 조항 변경 |
|
제52조 [감사의 직무] ① 감사는 회사의 회계와 업무를 감사한다. ② 감사는 회의의 목적사항과 소집의 이유를 기재한 서면을 이사회에 제 출하여 임시주주총회의 소집을 청구할 수 있다. ③ 감사는 그 직무를 수행하기 위하여 필요한 때에는 자회사에 대하여 영업의 보고를 요구할 수 있다. 이 경우 자회사가 지체없이 보고를 하지 아니할 때 또는 그 보고의 내용을 확인할 필요가 있는 때에는 자회사의 업무와 재산상태를 조사할 수 있다. ④ 감사에 대해서는 제40조 제3항의 규정을 준용한다. ⑤ 감사는 회사의 비용으로 전문가의 도움을 구할 수 있다. ⑥ 감사는 필요하면 회의의 목적사항과 소집이유를 적은 서면을 이사(소집권자가 있는 경우에는 소집권자)에게 제출하여 이사회 소집을 청구할 수 있다. ⑦ 제6항의 청구를 하였는데도 이사가 지체 없이 이사회를 소집하지 아니하면 그 청구한 감사가 이사회를 소집할 수 있다. |
제52조[감사위원회의 직무 등] ① 감사위원회는 회사의 회계와 업무를 감사한다. ② 감사위원회는 회의의 목적사항과 소집의 이유를 기재한 서면을 이사회에 제출하여 임시주주총회의 소집을 청구할 수 있다. ③ 감사위원회는 필요한 경우 회사의 비용으로 전문가의 조력을 구할 수 있다. ④ 감사위원회는 그 직무를 수행하기 위하여 필요한 때에는 자회사에 대하여 영업의 보고를 요구할 수 있다. 이 경우 자회사가 지체없이 보고를 하지 아니할 때 또는 그 보고의 내용을 확인할 필요가 있는 때에는 자회사의 업무와 재산상태를 조사할 수 있다. ⑤ 감사위원회는 제1항 내지 제4항 외에 이사회가 위임한 사항을 처리한다. ⑥ 감사위원회는 필요하면 회의의 목적사항과 소집이유를 적은 서면을 이사(소집권자가 있는 경우에는 소집권자)에게 제출하여 이사회 소집을 청구할 수 있다. ⑥ 제6항의 청구를 하였는데도 이사가 지체 없이 이사회를 소집하지 아니하면 그 청구한 감사위원회가 이사회를 소집할 수 있다. |
감사위원회 도입으로 인한 조항 변경 |
|
제53조 [감사의 감사록] 감사는 감사에 관하여 감사록을 작성하여야 하며, 감사록에는 감사의 실시요령과 그 결과를 기재하고 감사를 실시한 감사가 기명날인 또는 서명하여야 한다. |
제53조 [감사록] ① 감사위원회는 감사에 관하여 감사록을 작성하여야 한다 ② 감사록에는 감사의 실시요령과 그 결과를 기재하고 감사를 실시한 감사위원회 위원이 기명날인 또는 서명하여야 한다. |
감사위원회 도입으로 인한 조항 변경 |
|
제54조[감사의 보수와 퇴직금] ① 감사의 보수와 퇴직금에 관하여는 제41조의 규정을 준용한다. ② 감사의 보수를 결정하기 위한 의안은 이사의 보수결정을 위한 의안과 구분하여 상정, 의결하여야 한다. |
(삭제) | 감사위원회 도입으로 인한 조항 변경 |
□ 이사의 선임
가. 후보자의 성명ㆍ생년월일ㆍ추천인ㆍ최대주주와의 관계ㆍ사외이사후보자 등 여부
| 후보자성명 | 생년월일 | 사외이사 후보자여부 |
감사위원회 위원인 이사 분리선출 여부 |
최대주주와의 관계 | 추천인 |
|---|---|---|---|---|---|
| 정진섭 | 1967년 10월 4일 | 사외이사 후보 | - | - | 이사회 |
| 나경아 | 1964년 10월 3일 | 사외이사 후보 | 분리선출 | - | 이사회 |
| 총 ( 2 ) 명 | |||||
나. 후보자의 주된직업ㆍ세부경력ㆍ해당법인과의 최근3년간 거래내역
| 후보자성명 | 주된직업 | 세부경력 | 해당법인과의 최근3년간 거래내역 |
|
|---|---|---|---|---|
| 기간 | 내용 | |||
| 정진섭 | 대학교수 | 2009-현재 2014-현재 2020-2022 2008-2009 |
現.충북대 경영대학 교수 現.충북 투자유치 자문위원 現.충북 미래위원 및 미래비전자문단 前.성균관대 경영대학 연구교수 |
없음 |
| 나경아 | 대학교수 | 2020.9-현재 2004.12-2005.10 2001.4-2001.12 |
現.충북대 경영대학 교수 前.우리CA자산운용 상무 前.슈로더투자신탁운용 이사 |
없음 |
다. 후보자의 체납사실 여부ㆍ부실기업 경영진 여부ㆍ법령상 결격 사유 유무
| 후보자성명 | 체납사실 여부 | 부실기업 경영진 여부 | 법령상 결격 사유 유무 |
|---|---|---|---|
| 정진섭 | 해당사항 없음 | 해당사항 없음 |
해당사항 없음 |
| 나경아 | 해당사항 없음 |
해당사항 없음 |
해당사항 없음 |
라. 후보자의 직무수행계획(사외이사 선임의 경우에 한함)
| □ 정진섭, 나경아 후보자의 직무수행계획 당사 경영의 투명성 제고에 크게 이바지하고 경영사항의 의사결정을 위한 전문지식을 제공함으로써 당사의 이익증진을 위하여 노력할 예정입니다. |
마. 후보자에 대한 이사회의 추천 사유
| □ 정진섭 후보자에 대한 이사회의 추천 사유 후보자는 서울대학교 경영학과 석사/박사를 졸업하고, KOTRA 투자전략팀을 거쳐 성균관대학교 경영대학 연구교수로 재직한 바 있습니다. 충청북도 투자유치위원회 위원 등 다수의 전문기관에서 자문위원으로서의 업무 경험이 있는 바, 금번 2021.12 임시주주총회에서 당사의 사외이사 후보자 및 감사위원회 위원으로 추천되어 회사 발전에 크게 기여할 수 있을 것으로 판단되어 추천합니다. □ 나경아 후보자에 대한 이사회의 추천 사유 후보자는 Cambridge 대학 MBA 및 Alberta 대학에서 회계학을 전공하고, 슈로더투자신탁운용 회계 재무이사와 우리CA자산운용 재무담당 상무로 재직한 바 있으며 현재 충북대학교 국제경영학과 회계학 강의 및 연구교수로 재직 중에 있습니다. 금번 2021.12 임시주주총회에서 당사의 사외이사 후보자 및 감사위원회 위원으로 추천되어 회사 발전에 크게 기여할 수 있을 것으로 판단되어 추천합니다. |
확인서
 |
|
확인서_사외이사 정진섭_서명 |
 |
|
확인서_사외이사 나경아_서명 |
※ 기타 참고사항
해당사항 없음
□ 감사위원회 위원의 선임
가. 후보자의 성명ㆍ생년월일ㆍ추천인ㆍ최대주주와의 관계ㆍ사외이사후보자 등 여부
| 후보자성명 | 생년월일 | 사외이사 후보자여부 |
감사위원회 위원인 이사 분리선출 여부 |
최대주주와의 관계 | 추천인 |
|---|---|---|---|---|---|
| 김영준 | 1961년 1월 28일 | 사외이사 후보 | - | - | 이사회 |
| 정진섭 | 1967년 10월 4일 | 사외이사 후보 | - | - | 이사회 |
| 나경아 | 1964년 10월 3일 | 사외이사 후보 | 분리선출 | - | 이사회 |
| 총 ( 3 ) 명 | |||||
나. 후보자의 주된직업ㆍ세부경력ㆍ해당법인과의 최근3년간 거래내역
| 후보자성명 | 주된직업 | 세부경력 | 해당법인과의 최근3년간 거래내역 |
|
|---|---|---|---|---|
| 기간 | 내용 | |||
| 김영준 | 대학교수 | 20.06~현재 18.03~현재 01.03~현재 |
現.(주)레피다인 대표이사 現.프레스티지바이오로직스(주)사외이사 現.연세대학교 생화학과 교수 |
없음 |
| 정진섭 | 대학교수 | 2009-현재 2014-현재 2020-2022 2008-2009 |
現.충북대 경영대학 교수 現.충북 투자유치 자문위원 現.충북 미래위원 및 미래비전자문단 前.성균관대 경영대학 연구교수 |
없음 |
| 나경아 | 대학교수 | 2020.9-현재 2004.12-2005.10 2001.4-2001.12 |
現.충북대 경영대학 교수 前.우리CA자산운용 상무 前.슈로더투자신탁운용 이사 |
없음 |
다. 후보자의 체납사실 여부ㆍ부실기업 경영진 여부ㆍ법령상 결격 사유 유무
| 후보자성명 | 체납사실 여부 | 부실기업 경영진 여부 | 법령상 결격 사유 유무 |
|---|---|---|---|
| 김영준 | 해당사항 없음 | 해당사항 없음 |
해당사항 없음 |
| 정진섭 | 해당사항 없음 | 해당사항 없음 |
해당사항 없음 |
| 나경아 | 해당사항 없음 |
해당사항 없음 |
해당사항 없음 |
라. 후보자에 대한 이사회의 추천 사유
| □ 김영준 후보자에 대한 이사회의 추천 사유 후보자는 서울대학교 미생물학을 졸업하고, Stanford Univ.에서 미생물학 석사와 동 대학원에서 분자유전학박사를 취득하였습니다. 현재는 연세대학교에서 생화학과 교수로 재직 중에 있으며 다수의 전문기관에서 자문위원으로서의 업무 경험이 있는 바, 지난 2018.03. 주주총회에서 당사의 사외이사로 신규선임된 이후로 회사 발전에 크게 기여할 수 있을 것으로 판단하여 감사위원회 후보로 추천합니다. □ 정진섭 후보자에 대한 이사회의 추천 사유 후보자는 서울대학교 경영학과 석사/박사를 졸업하고, KOTRA 투자전략팀을 거쳐 성균관대학교 경영대학 연구교수로 재직한 바 있습니다. 충청북도 투자유치위원회 위원 등 다수의 전문기관에서 자문위원으로서의 업무 경험이 있는 바, 금번 2021.12 임시주주총회에서 당사의 사외이사 후보자 및 감사위원회 위원으로 추천되어 회사 발전에 크게 기여할 수 있을 것으로 판단되어 감사위원회 후보자로 추천합니다. □ 나경아 후보자에 대한 이사회의 추천 사유 후보자는 Cambridge 대학 MBA 및 Alberta 대학에서 회계학을 전공하고, 슈로더투자신탁운용 회계 재무이사와 우리CA자산운용 재무담당 상무로 재직한 바 있으며 현재 충북대학교 국제경영학과 회계학 강의 및 연구교수로 재직 중에 있습니다. 금번 2021.12 임시주주총회에서 당사의 사외이사 후보자 및 감사위원회 위원으로 추천되어 회사 발전에 크게 기여할 수 있을 것으로 판단되어 감사위원회 후보자로 추천합니다. |
확인서
 |
|
확인서_감사위원회위원 김영준_서명 |
|
|
|
확인서_사외이사 정진섭_서명 |
|
|
|
확인서_사외이사 나경아_서명 |
※ 기타 참고사항
해당사항 없음
IV. 사업보고서 및 감사보고서 첨부
가. 제출 개요
| 제출(예정)일 | 사업보고서 등 통지 등 방식 |
|---|---|
| - | - |
나. 사업보고서 및 감사보고서 첨부
-
※ 참고사항
|
□ 전자투표 및 전자위임장 권유에 관한 안내 |